Im Rahmen des Baden-Marathons am 25.09.16, nahm zwischen ungefähr 6000 Läufern die HHAC Labor Dr. Heusler GmbH bei dem integrierten Spendenmarathon (Business Team Marathon) mit zwei Laufteams teil. Es galt die Laufstrecke von 42 km, aufgeteilt auf 4 Team-Läufer mit 15, 9, 12 und 6 km zu bewältigen. Beide Teams bezwangen die Laufstrecke problemlos. Die gesammelten Spenden in Höhe von 1.500 € kommen dem Arbeitskreis Leben Karlsruhe e.V. (http://www.ak-leben.de) zugute, der mit Hilfe ehrenamtlicher Mitarbeiter Suizidgefährdete, deren Angehörige und Hinterbliebene betreut.
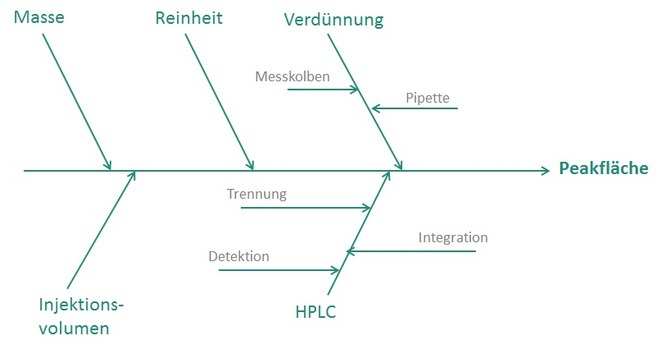
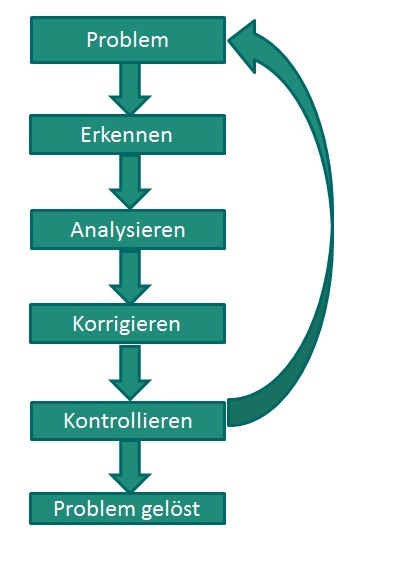




Kontakt
Sie haben eine Anfrage, die Sie gerne vorab mit uns klären möchten?
Dann rufen Sie uns gerne an. Für weitere Anfragen können Sie uns auch per E-Mail kontaktieren oder unser Kontaktformular nutzen.
Sie haben eine
Frage an uns?
Wir freuen uns auf Ihre Nachricht!
Kontakt
Sie haben eine Anfrage, die Sie gerne vorab mit uns klären möchten?
Dann rufen Sie uns gerne an. Für weitere Anfragen können Sie uns auch per E-Mail kontaktieren oder unser Kontaktformular nutzen.
Sie haben eine Frage an uns?
Wir freuen uns auf Ihre Nachricht!