Abstract
Essenzielle Voraussetzung für die erfolgreiche Durchführung von Stabilitätsstudien mit Betäubungsmitteln (BtM) ist, dass neben den Anforderungen von GMP und ICH auch die des Betäubungsmittelgesetzes vollumfänglich umgesetzt werden. Ein reibungsloser Projektablauf lässt sich mit einer klaren Strategie unter Einbezug eines Dienstleisters erreichen, der die erforderliche Kapazität für die BtM-Aufbewahrung, die Erlaubnis zum Umgang mit verschiedenen BtM-Stoffen und die entsprechende Expertise aufweist. Für den Sponsor resultieren daraus eine Einsparung enormer Investitionskosten, die Entlastung seines BtM-Verantwortlichen sowie eine deutliche Reduktion von BtM-Verkehr und Dokumentationsaufwand.
Einleitung
Stabilitätsprüfungen sowie generell analytische Prüfungen von Betäubungsmitteln (BtM) nehmen insofern eine Sonderstellung ein, da zusätzlich zu den GMP- und ICH-Anforderungen auch das Betäubungsmittelgesetz vollumfänglich beachtet werden muss. Beginnend mit der Erlaubnis zum Umgang mit BtM betrifft dies alle nachfolgenden Schritte im Prozessablauf von der Analytik über die Aufbewahrung bis hin zur Vernichtung der BtM-Muster. Beim Outsourcing von BtM-Projekten teilen sich die Aufgaben und auch die Verantwortlichkeiten zwischen Auftraggeber und Auftragnehmer auf. Abhängig vom Umfang des Outsourcings, d. h. Durchführung der Analytik mit oder ohne Einlagerung von Stabilitätsmustern, unterscheiden sich die an den Dienstleister zu stellenden Anforderungen v. a. in Bezug auf seine BtM-Erlaubnis für Stoffart und -menge sowie die baulichen/ sicherheitstechnischen Voraussetzungen für die Einlagerung von Mustern. Auf Sponsor-Seite lassen sich durch das Outsourcing von BtM-Stabilitätsprüfungen Investitionskosten für die Schaffung von Einlagerungskapazitäten vermeiden und zusätzlich wird sein BtM-Verantwortlicher erheblich entlastet. Sofern die Strategie gefahren wird, neben der Analytik auch die Klimaeinlagerung an den Outsourcing-Partner zu vergeben, reduzieren sich außerdem Ausmaß des BtM-Verkehrs und Dokumentationsaufwand beträchtlich. Für einen reibungslosen Projektverlauf ist die Wahl des geeigneten Outsourcing- Partners genauso wichtig wie ein Zeitmanagement, das entsprechende Zeitfenster z. B. für die Beantragung von Genehmigungen berücksichtigt.
Betäubungsmittel
Im ursprünglichen Sinn wurden als BtM diejenigen Stoffe bezeichnet, die zur Betäubung starker Schmerzen angewandt wurden. Beispielhaft werden hier Opium, Morphin und Kokain genannt. Heute ist im Betäubungsmittelgesetz (BtMG, [1]) eine klare Definition gegeben: Unter Betäubungsmitteln werden alle diejenigen Stoffe und Zubereitungen verstanden, die in Anlage I–III zu §1 Abs. 1 BtMG aufgelistet sind. Diese 3 Anlagen werden bei Bedarf aktualisiert. Sie teilen Betäubungsmittel in 3 grundsätzliche Gruppen ein. Unterschieden wird zwischen
- nicht verkehrsfähigen BtM (Anlage I),
- verkehrsfähigen aber nicht verschreibungsfähigen BtM (Anlage II) sowie
- verkehrsfähigen und verschreibungsfähigen BtM (Anlage III)
Anforderungen vor der Stabilitätsprüfung
Rechtliche Anforderungen zum Umgang mit BtM
Insbesondere zur Vermeidung einer missbräuchlichen Verwendung sowie einer potenziellen Suchtentstehung, ist der Umgang mit BtM gesetzlich festgelegt und im BtMG sowie den dazu erlassenen Verordnungen sehr streng geregelt. So ist als grundlegende Voraussetzung eine Erlaubnis zum Umgang mit BtM erforderlich für das:
- Anbauen,
- Herstellen,
- Handeln,
- Einführen,
- Ausführen,
- Abgeben,
- Veräußern,
- Inverkehrbringen,
- Erwerben.
Die Erlaubnis zum Umgang mit BtM muss beantragt werden und wird bei Erfüllung aller Voraussetzungen von der Bundesopiumstelle für eine Betriebsstätte und den benötigten Umfang erteilt. In dieser Erlaubnis wird auch der BtM-Verantwortliche des Unternehmens/der Einrichtung genannt, dessen Eignung durch ein Führungszeugnis und die entsprechende berufliche Qualifikation/Expertise zuvor belegt wurde. Die Funktion als BtM-Verantwortlicher ist alles andere als ein Ehrenamt: Als öffentlich-rechtlicher Garantenträger hat der BtM-Verantwortliche für den ordnungsgemäßen Umgang mit BtM Sorge zu tragen und kann bei einem Verstoß auch persönlich haftbar gemacht werden. Die Benennung eines BtM-Verantwortlichen ist zwingende Voraussetzung für den Erhalt einer Erlaubnis zum Umgang mit BtM und damit für die Teilnahme am BtM-Verkehr.
Lagerung und Aufbewahrung von BtM
Vor allem bei Stabilitätsprüfungen aber auch bei der Durchführung analytischer Qualitätskontrollen ist eine Lagerung bzw. kurzzeitige Aufbewahrung von BtM-Mustern erforderlich. Für die kurz- und langfristige Aufbewahrung von BtM sind explizite gesetzliche Vorgaben einzuhalten. Entsprechende Regelungen finden sich im BtMG sowie in Richtlinien der Bundesopiumstelle. Gemäß § 15 BtMG sind „BtM gesondert aufzubewahren und gegen unbefugte Entnahme zu sichern“. Grundsätzlich ist eine Aufbewahrung in Schränken oder in Räumen möglich, wobei die baulichen Anforderungen (z.B. Widerstandsgrad, Wanddicke) explizit in den Richtlinien vorgegeben sind. Abhängig von der Art der BtM sowie der aufzubewahrenden Menge ist eine elektrische Überwachung von Schränken (allseitig feldmäßig durch kapazitive Feldänderungsanlage) bzw. Räumen (durch Einbruchmeldeanlage mittels Körperschallprinzip) mit Alarmweiterschaltung zur Polizei erforderlich. Die sicherheitstechnischen Anforderungen sind in Abb. 1 zusammengefasst. Zur Ermittlung der Sicherungsmaßnahmen empfiehlt es sich, den Sicherungsrechner der Bundesopiumstelle auf der entsprechenden Webseite [2] zu nutzen und die Sicherungsmaßnahmen bereits in der Projektierungsphase mit der Bundesopiumstelle abzustimmen.
Vom Sender zum Empfänger – BtM auf dem Weg
Grundsätzlich ist zu unterscheiden, ob BtM lediglich innerhalb von Deutschland transportiert werden oder ob es sich um einen grenzüberschreitenden Transport handelt. Der grenzüberschreitende Transport ist geregelt in der Betäubungsmittel-Außenhandelsverordnung und ist deutlich aufwendiger zu organisieren als der innerdeutsche Transportweg. Beim Außenhandel ist der Ablauf für die Einfuhr des BtM durch den Empfänger und auch die Ausfuhr durch den Sender explizit geregelt [3]. Die einzuhaltenden Wege sind schematisch in Abb. 2 beschrieben. Für jede Einfuhr- und für jede Ausfuhrsendung muss eine Genehmigung bei der Bundesopiumstelle beantragt werden. Hierzu ist ein amtliches Formblatt zu verwenden, das gewissenhaft ausgefüllt werden muss. Der Prozess des Außenhandels mit BtM erfolgt mit den Hauptakteuren:
- inländische Behörde
- ausländische Behörde
- Einführer
- Ausführer
- ggf. Zoll in beiden betroffenen Ländern (entfällt bei EU-Warenverkehr)
Zunächst stellt der Einführer bei seiner inländischen Behörde (in Deutschland ist das die Bundesopiumstelle) einen Einfuhrantrag (siehe 1 in Abb. 2). Wird dieser genehmigt, so erhält er zwei Ausfertigungen einer Einfuhrgenehmigung, eine dritte Ausfertigung erhält die Behörde des Ausfuhrlandes (2). Der Einführer gibt eine Ausfuhrgenehmigung an den Ausführer weiter (3), der hiermit einen Ausfuhrantrag bei seiner Behörde stellen kann (4). Wird dieser genehmigt, so erhält der Ausführer zwei Ausfertigungen der Ausfuhrgenehmigung, eine dritte Ausfuhrgenehmigung erhält die Berhörde des Einfuhrlandes direkt von der ausländischen Behörde (5). Die Ausfuhrgenehmigungen werden bei Beteiligung des Zolls für die Ausfuhr an der Grenze benötigt (7). Ein Exemplar wird hier vom Zoll mit einem Vermerk versehen und muss vom Ausführer zusammen mit der Ausfuhranzeige an seine zuständige Berhörde versandt werden (11). Das beim Einführer bis dato noch verbliebene Exemplar der Einfuhrerlaubnis benötigt der Zoll des Einfuhrlandes für die Bearbeitung. Diese Einfuhrgenehmigung wird vom Zoll mit einem Vermerk versehen (8) und muss anschließend vom Einführer zusammen mit einer Einfuhranzeige an seine zuständige Behörde versandt werden (10). Durch den beschriebenen Prozess ist sichergestellt, dass alle am BtM-Verkehr beteiligten Akteure denselben Informationsstand haben und jederzeit nachvollziehbar ist, wo sich die BtM gerade befinden. Auch der Binnenhandel ist klar geregelt [4]. Hier ist ebenso einem vorgeschriebenen Ablaufplan zu folgen, der in Abb. 3 beschrieben ist. Es sind nur drei Akteure beteilig
- die Behörde,
- der Abgebende sowie
- der Erwerber.
Zentrales Dokument beim Binnenhandel ist der Abgabebeleg bestehend aus Abgabemeldung, Empfangsbestätigung, Lieferschein und Lieferscheindoppel. Der Abgebende füllt diesen Formularsatz aus und gibt zusammen mit den BtM eine Empfangsbestätigung sowie einen Lieferschein an den Erwerber ab (siehe 1 in Abb. 3). Die Bundesopiumstelle erhält parallel dazu eine Abgabemeldung von ihm (2). Der Erwerber bestätigt dann den Erhalt der BtM, indem er die Empfangsbestätigung an den Abgebenden zurückgibt (3). Falls Abweichungen zwischen Dokumentation und tatsächlicher Lieferung festgestellt werden, so müssen diese vom Erwerber auf dem Lieferschein sowie der Empfangsbestätigung vermerkt und vom Abgebenden auf dem Lieferscheindoppel notiert werden. Durch Zusendung des Lieferscheindoppels durch den Abgebenden wird die Bundesopiumstelle über die Korrektur informiert (4). Mit diesem Prozedere wird wiederum sichergestellt, dass jederzeit nachvollzogen werden kann, wo sich die BtM zum aktuellen Zeitpunkt befinden.
Anforderungen während der Stabilitätsprüfung
Gemäß §17 BtMG sind Aufzeichnungen zum aktuellen Bestand der BtM zwingend vorgeschrieben. Unverzüglich nach jeder Änderung muss der aktuelle Bestand an BtM, für jeden Zu- bzw. Abgang, getrennt für jedes BtM sowie jede Betriebsstätte, dokumentiert werden. Diese Dokumentation muss gesondert für 3 Jahre aufbewahrt werden und ist ggf. inspektionsrelevant. Darüber hinaus sind halbjährlich für jede Betriebsstätte, getrennt für jedes BtM, unaufgefordert Bestandsmeldungen gemäß § 18 BtMG an die Bundesopiumstelle zu machen.
Anforderungen nach der Stabilitätsprüfung
Die nach Abschluss der Stabilitätsprüfung nicht mehr verwendbaren BtM-Muster müssen gemäß den Vorschriften von § 16 BtMG entsprechend vernichtet werden. Welche Methode hierfür eingesetzt werden kann, ist nicht vorgeschrieben, wohl aber, dass die Vernichtung so zu erfolgen hat, dass eine Rückgewinnung – auch teilweise – unmöglich ist. Die dokumentierte Vernichtung erfolgt im 6-Augen-Prinzip, sodass neben dem Durchführenden auch zwei Zeugen anwesend sind. Der Schutz von Mensch und Umwelt ist dabei selbstverständlich zu beachten. Für das Vernichtungsprotokoll besteht eine Aufbewahrungspflicht von drei Jahren.
Voraussetzungen für das Outsourcing
Um Stabilitätsprüfungen von Betäubungsmitteln an einen Dienstleister auslagern zu können, sind gegenüber dem Outsourcing von Nicht-BtM-Prüfungen einige zusätzliche Anforderungen zu erfüllen. Grundlegende Voraussetzung ist die Erlaubnis zum Umgang mit BtM, die beim Dienstleister für die entsprechenden BtM und für den entsprechenden Umfang vorliegen muss. In der Planungsphase ist bereits die Entscheidung zu treffen, ob beim Outsourcing-Partner im Rahmen der Stabilitätsprüfung lediglich die Analytik zu den entsprechend vorgegebenen Zeitpunkten erfolgen oder ob bei diesem auch gleichzeitig die Einlagerung der Muster beauftragt werden soll. Werden die Stabilitätsmuster beim Dienstleister eingelagert, so wirkt sich dies massiv auf dessen BtM-Bestand aus. Die Aufbewahrung größerer BtM-Mengen erfordert hohe Sicherheitsmaßnahmen, deren Aufbau mit erheblichen Investitionen verbunden ist und auch einige Bauzeit in Anspruch nimmt. Werden alle sicherheitstechnischen und baulichen Anforderungen erfüllt, kann der Dienstleister eine an Art und Menge der BtM angepasste Erlaubnis beantragen. Ein entsprechendes Zeitfenster als Vorlauf vor Beginn der Stabilitätsstudie sollte hierfür eingeplant werden. Während es zwar einige Dienstleister gibt die BtM analytisch prüfen können, gibt es nur wenige, die auch die Stabilitätseinlagerung übernehmen und damit sowohl die Bereitschaft besitzen, in die erforderliche Infrastruktur zu investieren als auch eine deutlich größere Verantwortung im BtM-Verkehr zu übernehmen.
Starke Kombination aus Kapazität und Flexibilität – ein Praxisbeispiel
Um als Dienstleister Stabilitätsprojekte mit BtM zuverlässig und routiniert durchführen zu können, sind Flexibilität und Kapazität unerlässlich. Flexibilität bezieht sich hier auf die einzelnen BtM-Stoffe, für die eine Umgangserlaubnis vorliegt bzw. beantragt werden kann. Kapazität betrifft in erster Linie den für die Einlagerung zur Verfügung stehenden Platz. Die einfachste und damit auch kostenintensivste Variante ist es, einen Wertschutzraum zur Verfügung zu stellen, in dem die entsprechenden Klimaschränke oder Klimaräume untergebracht sind. Dieser Wertschutzraum muss den aktuellen baulichen und sicherheitstechnischen Anforderungen der Bundesopiumstelle entsprechen. Hier empfehlen sich dringend die Benutzung des Sicherungsrechners sowie die Abstimmung mit der Bundesopiumstelle bereits in der Projektierungsphase. Für diesen Weg hat sich HHAC Labor Dr. Heusler entschieden und einen solchen Wertschutzraum errichtet. Einen Einblick in den Wertschutzraum gibt Abb. 4. Dieser Wertschutzraum bietet hinreichend Platz, um mehrere Klimaschränke mit den entsprechend geforderten Temperatur-/Feuchte-Bedingungen unterzubringen. Alle Klimaschränke sind an ein qualifiziertes Messwerterfassungssystem mit periodischer Speicherung der Messwerte angeschlossen. Ebenso ist eine kurzzeitige Aufbewahrung von Betäubungsmitteln in diesem Wertschutzraum bei Raumtemperatur möglich. Diese erfolgt in einem separaten dafür vorgesehenen Tresor. Vor unbefugtem Zutritt wird der Raum mithilfe von Körperschallmeldern überwacht, die bei Auslösung eine direkte Alarmierung der Polizei veranlassen, die dann innerhalb weniger Minuten vor Ort ist. Die Zutrittsberechtigung ist nur wenigen autorisierten Personen gestattet, die dazu im Besitz mehrerer Kennwörter sowie eines Chip-Schlüssels sind. Der Wertschutzraum erfüllt die höchsten beschriebenen baulichen und sicherheitstechnischen Anforderungen. Von daher können sowohl unterschiedliche BtM-Stoffe als auch größere Mengen hierin aufbewahrt werden. Abgestimmt an das vom Kunden beauftragte Stabilitätsprojekt ist ggf. eine Anpassung der BtM-Erlaubnis erforderlich, die aber zeitnah erfolgt, da alle baulichen und sicherheitstechnischen Anforderungen vollumfänglich erfüllt sind und der höchsten Sicherungsstufe entsprechen.
Vorteile des Outsourcings für den Sponsor
Die Übertragung von Aufgaben an einen Dienstleister schafft neue Kapazitäten, die genutzt werden, um sich auf seine Kernkompetenzen konzentrieren zu können. Dies hat somit positive Auswirkung auf die eigene Wettbewerbsfähigkeit [5]. Ein großer Vorteil des Outsourcings für den Sponsor ist, dass damit nicht nur eine Übernahme von Aufgaben durch den entsprechenden Dienstleister erfolgt, sondern damit auch eine Übernahme von Verantwortung verbunden ist. Gerade beim Umgang mit BtM ist dies eine nicht zu unterschätzende Erleichterung für den BtM-Verantwortlichen beim Auftraggeber. Verantwortlich für die Bereitstellung der notwendigen Kapazität ist der Dienstleister, der dafür Sorge tragen muss, dass er neben den GMP-Anforderungen die gesetzlich festgelegten Forderungen des BtMG vollumfänglich erfüllt. Wenn neben der analytischen Prüfung der Stabilitätsmuster auch die Einlagerung beim Dienstleister erfolgt, entfällt das wiederholte Abgabe-/Erwerb- bzw. Ausfuhr-/Einfuhr-Prozedere vom Sponsor zum Dienstleister. Sämtliche Dokumentationen während der Stabilitätsprüfung werden vom Dienstleister übernommen und dieser ist auch für die unaufgefordert abzugebenden Meldungen an die Bundesopiumstelle verantwortlich. Nach Abschluss der Stabilitätsprüfung werden die verbliebenen Muster nach dem festgelegten und dokumentierten Ablauf vom Dienstleister vernichtet. Viele Aufgaben, die beim Umgang mit BtM zu erledigen sind, werden im Rahmen des Outsourcings vom BtM-Verantwortlichen des Dienstleisters übernommen und dieser trägt auch Sorge für den gesetzeskonformen Projektablauf im eigenen Unternehmen. Der Betäubungsmittelverantwortliche des Sponsor-Unternehmens wird dadurch ganz erheblich entlastet, was sich auch vereinfachend auf das firmeninterne Risikomanagement auswirkt [6]. Eine Zusammenfassung der Vorteile zeigt Abb. 5.
Fazit
Bei Stabilitätsprüfungen von BtM sind zusätzlich zu den Vorgaben von ICH und GMP auch die gesetzlichen Vorgaben des Betäubungsmittelgesetzes sowie die der dazu erlassenen Verordnungen zu erfüllen. Insbesondere zur Erfüllung der rechtlichen Anforderungen sind erhebliche Investitionen erforderlich, die umso größer sind, je mehr Flexibilität und Kapazität verfügbar sein muss. Durch das Outsourcing von BtM-Stabilitätsprüfungen werden diese Investitionskosten beim Sponsor vermieden und sein BtM-Verantwortlicher wird ganz erheblich entlastet. Sofern die Strategie gefahren wird, neben der Analytik auch die Klimaeinlagerung an den Outsourcing-Partner zu vergeben, reduzieren sich Ausmaß des BtM-Verkehrs und Dokumentationsaufwand nochmals erheblich. Um einen reibungslosen Projektverlauf zu gewährleisten, sind drei Fragestellungen zu klären:
- Erfolgt lediglich die Analytik oder zusätzlich auch die Einlagerung der BtM beim Dienstleister?
- Welcher Dienstleister ist geeignet (Kapazität, Flexibilität)?
- Welche Zeitfenster (für bauliche Maßnahmen, Erlaubnisanträge) müssen eingeplant werden?
Literatur
[1] Gesetz über den Verkehr mit Betäubungsmitteln (BtMG) Fassung vom 16.06.2017.
[2] www.bfarm.de/DE/Bundesopiumstelle/_node.html
[3] Betäubungsmittel-Außenhandelsverordnung (BtMAHV) Fassung vom 06.03.2017.
[4] Betäubungsmittel-Binnenhandelsverordnung (BtMBinHV) Fassung vom 17.08.2011.
[5] Eichhorn J. Outsourcing von Analytik in der Pharmaindustrie. Pharm. Ind. 2015;77(5):734-738
[6] Krebsbach et al. Risiko- und Chancenmanagement im Labor. Pharm. Ind. 2017;79(6):870-875.
Abbildung 1

Abbildung 2
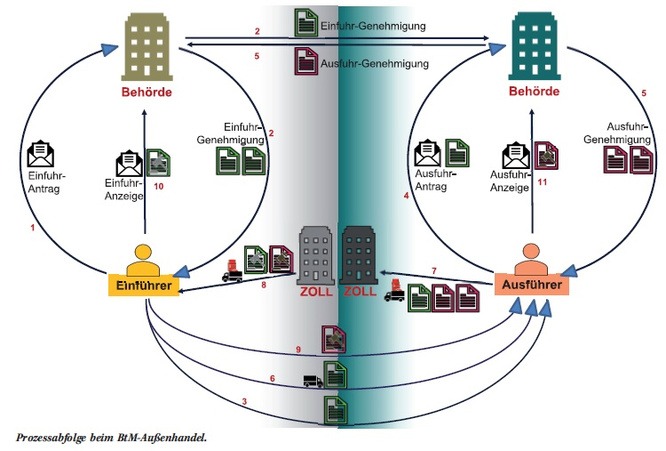
Abbildung 3
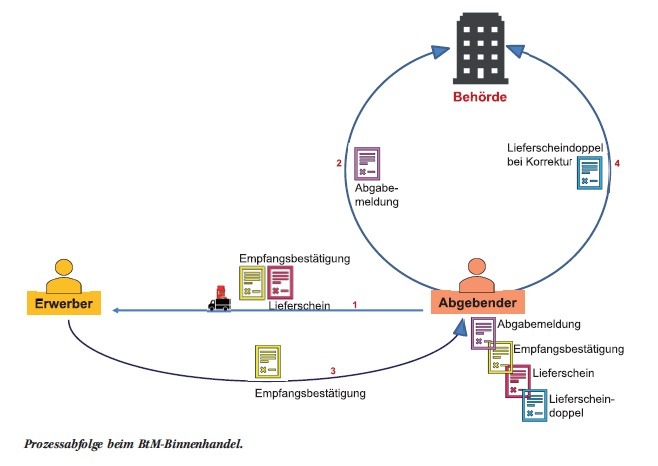
Abbildung 4

Abbildung 5


