Wie lässt sich die neue Akkreditierungs-Forderung der ISO 17025 in der Praxis umsetzen?
Die Beschäftigung mit Risiken und Chancen ist täglich gelebte Praxis in allen Unternehmen und fundamentale Voraussetzung, um am Markt zu bestehen. Während insbesondere größere Kapitalgesellschaften rechtlich längst zum kontinuierlichen Betreiben eines Risikomanagementsystems verpflichtet sind, betrifft die Forderung nach einem risikobasierten Ansatz nun auch alle Qualitätskontroll-/Prüflabore, die gemäß ISO 17025 akkreditiert sind und damit gleichzeitig die Anforderungen der ISO 9001 erfüllen müssen. Für kleinere Labore ist ein ausgefeiltes Risikomanagementsystem überdimensioniert und auch gar nicht erforderlich. Das Risikomanagement sollte hier den Blick auf das Wesentliche, die Konformität von Prüfergebnissen sowie die Kundenzufriedenheit, fokussieren und bringt damit einen echten Nutzen für das eigene Unternehmen.
Der Oberbegriff „Risikomanagementsystem“ bezieht sich auf mehr als nur auf die Konformität und Korrektheit von Ergebnissen, Verfahren und Methoden der durchgeführten Prüfungen und Analysen. Mit einem Risikomanagementsystem soll erreicht werden, dass Unternehmen vorbereitet in die Zukunft gehen, indem sie bestandsbedrohende Risiken rechtzeitig erkennen und darauf reagieren, Chancen nutzen können und so den erfolgreichen Fortbestand des Unternehmens sicherstellen. Insbesondere Aktiengesellschaften wird sofort klar sein, was sich hierunter verbirgt, sind sie doch rechtlich dazu verpflichtet, ein im Managementsystem verankertes Risikomanagement kontinuierlich zu betreiben und zu dokumentieren. Für kleinere Unternehmen, wie kleinen Eigentümer-geführten Firmen, hingegen ist ein ausgefeiltes Risikomanagementsystem ohne Frage überdimensioniert. Das Risikomanagementsystem sollte den Tätigkeiten und der Größe des Unternehmens angepasst sein. Mit der Forderung nach dem Management von Risiken und Chancen werden in Kürze alle Qualitätskontroll-/Prüflabore, die eine ISO 17025-Akkreditierung aufrechterhalten wollen, konfrontiert werden. Die neueste Version dieser ISO-Norm, deren In-Kraft-Treten noch in diesem Jahr erwartet wird, fordert explizit die Evaluierung und Bewertung von Risiken und Chancen sowie die Integration ins Managementsystem. Bislang gab es lediglich die Forderung nach „Vorbeugenden Maßnahmen“: „…Notwendige Verbesserungen und mögliche Fehlerquellen, entweder technischer Art oder bezüglich des Managementsystems, müssen ermittelt werden. Wenn Verbesserungsmöglichkeiten aufgezeigt werden oder wenn eine vorbeugende Maßnahme erforderlich ist, müssen Pläne für Maßnahmen entwickelt, umgesetzt und überwacht werden, um die Wahrscheinlichkeit für das Auftreten solcher Fehler zu verringern und Verbesserungsmöglichkeiten zu nutzen…“ [1]. Neu ist nun die Anforderung, dass das Labor „Maßnahmen plant und umsetzt, mit denen Risiken und Chancen behandelt werden … Das Laboratorium ist dafür verantwortlich zu entscheiden, welche Risiken und Chancen behandelt werden müssen…“ [2]. Die Forderung der ISO 17025 nach einem Risikomanagement war nach der Revision der ISO 9001:2015 zu erwarten, die die Beschäftigung mit Risiken und Chancen aufgegriffen hat. Für alle ISO 17025-akkreditierten Labore wird die Betrachtung von Chancen und Risiken nun verbindlich. Der Gedanke, sich mit Risiken und Chancen zu beschäftigen, ist dabei nicht neu. Bestehende gesetzliche Grundlagen fordern den Umgang mit Chancen und Risiken schon seit längerer Zeit, z. B. mit Neueinführung von § 91 Abs. 2 AktG (Aktiengesetz) im Rahmen des Gesetzes zur Kontrolle und Transparenz im Unternehmensbereich (KonTraG). Kapitalgesellschaften müssen ein Risikomanagementsystem installieren, nutzen, dokumentieren und eine Interne Revision einrichten. Zudem lässt sich die Forderung nach einem Risikomanagement auch aus den Sorgfaltspflichten eines GmbH-Geschäftsführers (§ 43 Abs. 1 GmbHG) ableiten. Vorschriften, wie genau das zu erfolgen hat, gibt es in beiden Gesetzestexten hingegen nicht. Ein Unternehmen hat damit einen großen Freiheitsgrad in der Umsetzung, kann bestehende hausinterne Strukturen dabei nutzen und die eigenen Bedürfnisse berücksichtigen. Alle Unternehmen -große wie kleine- betreiben bereits Tag für Tag ein aktives Risikomanagement. Die gelebte Praxis sieht so aus, dass Entscheidungen doch immer risikobasiert getroffen werden; eine wesentliche Grundlage, um am Markt erfolgreich zu sein. Das gilt für den operativen Bereich genauso wie für den strategischen. Der Umfang der Dokumentation dieser risikobasierten Entscheidungen unterscheidet sich zwischen großen und kleinen Unternehmen jedoch stark. Dieser Unterschied ist aufgrund der Tragweite von Entscheidungen -vor allem bei strategischen- und der personell zur Verfügung stehenden Kapazitäten auch durchaus gerechtfertigt. Unternehmen, die ein Risikomanagementsystem für sich angenommen und etabliert haben, werden ihre Risikobewertung nach intern festgelegten Regeln und den Grundsätzen eines ordentlichen Geschäftsführers ausführen.
Wie kann und sollte ein Labor, das nicht als Konzern/Aktiengesellschaft aufgestellt ist, mit der Forderung der ISO-Normen nach einem Risikomanagementsystem nun umgehen, d. h. wie kann die Umsetzung in der Praxis erfolgen und dabei auch noch einen echten Mehrwert für das eigene Unternehmen liefern?
Risiken und Chancen
Zunächst einmal muss definiert werden, was hier unter Risiken und Chancen verstanden werden soll:
Risiko
potentielles Eintreten eines Ereignisses, dass sich negativ auf das Erreichen der Unternehmensziele auswirkt Risiken sind in allen Systemen, Prozessen und Funktionen vorhanden, z. B. können einzelne Prozessschritte von den Vorgaben abweichen, es gibt Risiken beim Einkauf, bei der Lagerung, bei der Prüfung, bei der Beratung, … . Im Fokus steht dabei immer, was das Erreichen der Managementsystemziele Konformität und Kundenzufriedenheit gefährden kann. Das Schadensausmaß bei Eintritt kann dabei von einem geringen finanziellen Schaden bis hin zu weitreichenden unternehmensbedrohenden Konsequenzen reichen.
Für Chancen gelten dieselben Prinzipien, allerdings durchweg im positiven Sinn:
Chance
potentielles Eintreten eines Ereignisses, dass sich positiv auf das Erreichen der Unternehmensziele auswirkt Voraussetzung für die Erkennung und den Umgang mit Risiken ist eine risikobasierte Denkweise. Diese zeichnet sich durch zukunftsorientiertes und vorbeugendes Denken aus, hat potentielle Einflussfaktoren auf die Erreichung von Zielen sowie Eintrittswahrscheinlichkeiten und das Schadensausmaß im Blick, führt zur Entwicklung von Szenarien und auch dazu, dass das Eingehen von Risiken bewusst und in voller Absicht geschieht. Oberstes Ziel ist es, rechtzeitig einen potentiellen Einfluss zu erkennen, Chancen zu nutzen und Risiken zu steuern, immer mit Blick auf die Ziele des Managementsystems. Risikobasiertes Denken ist als kontinuierlicher Prozess und insbesondere als Bestandteil des kontinuierlichen Verbesserungsprozesses (KVP) zu verstehen.
Prinzipieller Aufbau eines Risikomanagementsystems
Die Betrachtung von strategischen, operativen, finanziellen und Marktumfeld-Risiken stellt den Mindestumfang des Risikomanagementsystems dar. Hierzu ist im Unternehmen die Erfassung der Risiken und der weitere Umgang damit zu organisieren. Der Umgang mit Risiken folgt dem PDCA-Zyklus mit den Bausteinen Plan, Do, Check und Act (s. Abb. 1). Ein erfolgreiches Risikomanagementsystem lebt davon, dass das Risikobewusstsein auf allen Unternehmensebenen vorhanden ist, d. h. alle Ebenen an einem Strang und in die gleiche Richtung ziehen. Ist diese Grundvoraussetzung geschaffen, kann mit der Identifizierung der Risiken begonnen werden. Die ermittelten Risiken werden nun einer Bewertung unterzogen, ein Steuerungsmechanismus sowie die zukünftige Kontrolle werden festgelegt. Von den verschiedenen Möglichkeiten, ein Risikomanagement zu betreiben und dies in die Unternehmensstrategie einzugliedern, wird hier beispielhaft die SWOT-Analyse ausgewählt, die aus den 3 Schritten besteht:
1. Stärken und Schwächen des Unternehmens mit Hilfe einer Unternehmensanalyse bestimmen
2. Chancen und Risiken aus der Umwelt-/Markt- und Mitbewerberanalyse ableiten [3]
3. Zusammenfassende Betrachtung der Stärken, Schwächen, Chancen und Risiken [4]
Im ersten Schritt werden die aktuellen und zukünftigen Stärken und Schwächen des Unternehmens ermittelt [5]. Damit sind z.B. kritische Erfolgsfaktoren oder die Position im Wettbewerbs-Vergleich gemeint. Zu betrachten sind [6]:
- Unternehmensstrategie
- Ad hoc-Auflistung wesentlicher vergangener Erfolge und Misserfolge
- Analyse der zentralen Ursachen für die wesentlichen Erfolge und Misserfolge
- Ad hoc-Auflistung der möglichen Schlüsselfaktoren für die künftige Entwicklung des Unternehmens, Prüfung auf Plausibilität, Bewertung der Schlüsselfaktoren für die Unternehmensentwicklung
- Ad hoc-Auflistung der Stärken und Schwächen für die Existenzsicherung des Unternehmens, die sich aus den Schlüsselfaktoren der künftigen Entwicklung ergeben können
- Bewertung der Stärken und Schwächen in Bezug auf die bedeutenden Wettbewerber
Im zweiten Schritt werden die Risiken und Chancen ermittelt. Dies soll vollständig, systematisch sowie kontinuierlich geschehen und setzt fundierte Kenntnis über das gesamte Unternehmen voraus. Hierzu eignet sich die PESTEL-Analyse [4, 7], bei der das politische, ökonomische, gesellschaftliche, technologische, ökologische und gesetzliche Umfeld betrachtet wird (s. Abb. 2). Das Ziel ist hier die Einschätzung der Attraktivität des Marktes, der Entwicklung des Marktes sowie der zukünftigen Einflussgrößen. Zusätzlich sollte auch das Wettbewerbsumfeld, d. h. Branche und Mitbewerber, betrachtet werden. Hinzu kommen dann noch die operativen Risiken des eigenen Unternehmens. Im Anschluss an die Identifizierung der Risiken folgt deren Bewertung anhand der beiden Parameter „Eintrittswahrscheinlichkeit“ und „Schadensausmaß“. Beide Parameter könnten mathematisch ausgedrückt werden, in der Praxis reicht aber eine qualitative Beschreibung in „fast sicher“ … “unwahrscheinlich“ (Eintrittswahrscheinlichkeit) bzw. „unbedeutend“ … “existenziell“ (Schadensausmaß) völlig aus. Werden alle Risiken anhand der beiden Parameter „Eintrittswahrscheinlichkeit“ und „Schadensausmaß“ geordnet, entsteht das Risiko-Portfolio (Abb. 3.). Auf einen Blick sind Bedrohungspotentiale hier sofort zu erkennen. Da die Risiken und deren Tragweiten nun bestimmt sind, erfolgt im nächsten Schritt die Steuerung der Risiken mit den zur Verfügung stehenden Maßnahmen (Abb. 4). Die Auswahl einer geeigneten Steuerungsmaßnahme hängt von der Positionierung des Risikos in der Matrix (Abb. 5) ab.
Im dritten Schritt werden nun die Stärken, Schwächen, Chancen und Risiken zusammenfassend betrachtet und in einer SWOT-Matrix angeordnet [4, 5, 7]. Hieraus entwickelt dann das Unternehmen die geeignete strategische Option.
Ein gelebtes Risikomanagement zeichnet sich durch eine kontinuierliche Kontrolle und Steuerung und damit einem Verbesserungsprozess aus. Hier ist es zunächst einmal wichtig festzulegen, wer welche Verantwortung im Prozessverlauf übernimmt. Um die Kontrolle dann möglichst einfach und präzise zu gestalten, werden Frühwarnindikatoren inklusive deren Soll- und Grenzwerten definiert. Im Fall von Überschreitungen muss dann mit Steuerungsmaßnahmen umgehend reagiert werden (Abb. 6).
Konsequenzen für das Qualitätskontroll-/Prüflabor
Die Etablierung eines komplexen, umfassenden Risikomanagementsystems ist für kleinere Labore überdimensioniert, nicht gefordert und auch gar nicht nötig [9, 10]. Es ist allerdings hilfreich, die einzelnen Stufen des oben beschriebenen Prozesses einmal zu verstehen, um die Einordnung in die Unternehmensstrategie realisieren zu können und um einschätzen zu können, auf welche Positionen im Prozess fundiert verzichtet werden kann. Anschließend reicht dann eine vereinfachte Umsetzung des Risikomanagementsystems völlig aus. Forderungen aus der ISO-Norm nach konkret anzuwendenden Methoden für den Risikoermittlungs- und Bewertungsprozess sind genauso wenig vorhanden wie die Vorgabe eines formalen Systems. Weiterhin gibt es keine Forderung, sich um alle Risiken sofort zu kümmern. Risiken sollten auch nicht grundsätzlich vermieden werden, weil damit schlussendlich auch Chancen ungenutzt bleiben könnten. Vielmehr stellt die Beschäftigung mit Risiken und Chancen eine separate Komponente des Managementsystems dar und ist nicht mehr eingebettet in die bislang bekannte Forderung nach vorbeugenden Maßnahmen. Die Verantwortung für das Risikomanagementsystem liegt beim Top-Management. Das systematische Managen von Risiken und Chancen muss in allen beteiligten Köpfen verankert sein, so dass Auswirkungen betrachtet werden in Bezug auf die Fähigkeit der Organisation, fortlaufend konforme (in Bezug auf Kunden, Gesetze, Behörden) Dienstleistungen zu erbringen. Das Unternehmen bestimmt die Risiken/Chancen selbst. Umwelt-Risiken (siehe oben PESTEL-Analyse) können hier außen vor bleiben, die Konzentration auf strategische und operative Risiken reicht zunächst einmal völlig aus.
Starthilfe für die Praxis
Wie bei jedem Projekt ist die eigene Grundhaltung entscheidend. Nur wer selbst voll und ganz hinter einem Projekt steht und dafür brennt, kann andere dafür begeistern. Zuerst gilt es daher, sich selbst die Sinnhaftigkeit des Umgangs mit Risiken und Chancen bewusst zu machen, ein Risikobewusstsein zu entwickeln. Daran schließt sich die Zusammenstellung einer Arbeitsgruppe, der RICH-Gruppe (RIsiken/CHancen) an, in der alle Bereiche des Unternehmens vertreten sind, z.B. Qualitätsmanagement, operativer Leistungsbereich, Einkauf, Buchhaltung, Personal, Marketing, Geschäftsleitung. Von den Mitgliedern der RICH-Gruppe wird eine offene Grundhaltung gegenüber Chancen und Risiken erwartet. Für einen gelungenen Start bieten sich zwei unterschiedliche Herangehensweisen an:
Methode A:
Die einzelnen Mitglieder der RICH-Gruppe ermitteln für ihren Aufgabenbereich die 5 wichtigsten Risiken und die 5 wichtigsten Chancen. Diese zunächst einmal subjektive Einschätzung wird in der RICH-Gruppe überprüft, diskutiert und abgestimmt. Am Ende wird eine Auflistung der Risiken und Chancen erhalten, inklusive Eintrittswahrscheinlichkeiten und Schadensausmaß sowie festgelegten Maßnahmen. Für jedes Risiko bzw. für jede Chance wird anschließend ein Verantwortlicher festgelegt.
Methode B:
Um einen leichteren Einstieg bei der Ermittlung von Chancen und Risiken zu finden, bietet sich eine Inventur an. Hier werden eingeleitete Maßnahmen zusammen mit dem Grund der Einführung aufgelistet. Der Grund der Einführung steht entweder für ein Risiko oder eine Chance. Diese Inventur sollte sich nur auf einen kurzen Zeitraum von z. B. einem Jahr beziehen.
Im Anschluss daran -und das ist beiden Methoden wieder gemeinsam- erfolgt eine periodische Fortführung der Beschäftigung mit Risiken. Das schließt jährliche/halbjährliche Treffen der RICH-Gruppe ein sowie einen jährlich erstellten Risikomanagementbericht als Bestandteil der Managementbewertung. Die Ermittlung von Risiken und Chancen, deren Bewertung sowie Maßnahmen und Verantwortliche können übersichtlich in Form einer Tabelle dargestellt werden. Diese Tabelle dient als to do-Liste und wird kontinuierlich fortgeschrieben (Abb.7).
Vorteile für das Labor
Da jedes Unternehmen tagtäglich risikobasiert handelt, um auf dem Markt zu bestehen, könnte bei rein oberflächlicher Betrachtung der neuen ISO-Forderung der Gedanke entstehen, dass lediglich zusätzliche Arbeit generiert wird ohne Einfluss auf den geschäftlichen Erfolg zu nehmen. Dabei führt die Erfüllung der Anforderungen insbesondere dazu, den Prozess der Beschäftigung mit Risiken und Chancen klar zu strukturieren sowie zu dokumentieren, wodurch die Unternehmensführung bei der Erreichung ihrer Ziele wirksam unterstützt wird. So wird durch die Betrachtung von Risiken und Chancen umfassendes Know-how gewonnen, die Planungssicherheit wird verbessert und Chancen werden nach Abwägung aktiv angegangen. Vorbeugen wird zur Gewohnheit und greift weiter als die vorbeugenden Maßnahmen, wie sie in der aktuellen Version der ISO-Norm beschrieben sind. Das Unternehmen geht vorbereitet in die Zukunft, überwacht bestandsbedrohende Risiken nachvollziehbar, erkennt sie frühzeitig und reduziert das Ausmaß. Ein optimales Verhältnis von Chancen und Risiken fördert nicht nur eine positive Unternehmenskultur, sondern unterstützt die Sicherstellung einer qualitativen Gleichmäßigkeit der Dienstleistung und sorgt für erhöhte Kundenbindung, -zufriedenheit und ein begründetes -vertrauen.
Fazit
Risikomanagement bzw. der Ansatz des risikobasierten Denkens ist nichts Neues. Die Forderung aus den ISO-Normen 17025 bzw. 9001 lassen sich auch von kleineren Qualitätskontroll-/Prüflaboren vollumfänglich einhalten. Die Etablierung eines ausgefeilten Risikomanagements ist dabei weder gefordert noch sinnvoll. Mit Blick auf das Wesentliche bringt die Beschäftigung mit Risiken und Chancen einen echten Nutzen für das Unternehmen und dessen Auftraggeber. Im Vordergrund steht dabei eine proaktive Unternehmenskultur mit zukunftsorientierter Fokussierung.
Literatur
[1] ISO/IEC 17025:2005. Allgemeine Anforderungen an die Kompetenz von Prüf- und Kalibrierlaboratorien
[2] ISO/IEC DIS 17025:2016. Allgemeine Anforderungen an die Kompetenz von Prüf- und Kalibrierlaboratorien
[3] Graumann M. Unternehmensverfassung, Strategie und Planung. Koblenz: ZFH; 2009.
[4] Bamberger I, Wrona T. Strategische Unternehmensführung. Strategien, Systeme, Methoden, Prozesse. München: Vahlen; 2012
[5] Corsten H. Grundlagen der Wettbewerbsstrategie. Stuttgart: Teubner; 1998
[6] Graumann M. Controlling. Begriff, Elemente, Methoden und Schnittstellen. Düsseldorf: IDW-Verlag; 2008.
[7] Jung R/ Bruck J/ Quarg S. Allgemeine Managementlehre. Lehrbuch für die angewandte Unternehmens- und Personalführung. Berlin: Schmidt; 2011
[8] Meier H. Unternehmensführung. Aufgaben und Techniken betrieblichen Managements; Unternehmenspolitik und strategische Planung, Unternehmensplanung und Organisation, Human-Resources-Management. Herne: NWB; 2010
[9] ISO-Komitee. Risk-based thinking in ISO 9001:2015. ISO/TC 176/SC 2/N1284
[10] ISO-Komitee. ISO 9001:2015 Risk-based thinking. ISO/TC 176/SC 2/N1283
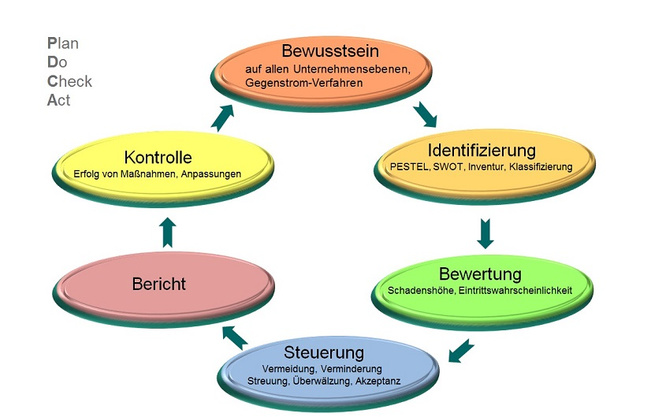
Abbildung 1: PDCA-Zylkus (PDCA: Plan, Do, Check, Act)
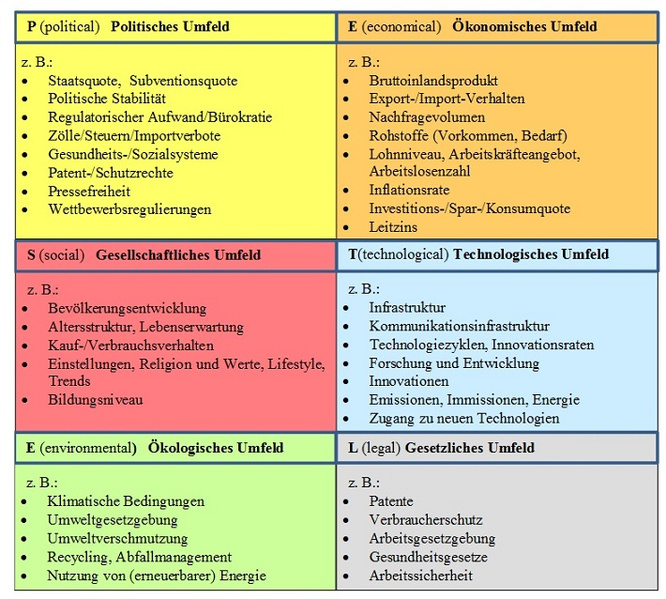
Abbildung 2: Einflussgrößen der PESTEL-Analyse in Anlehnung an [3,6,8]
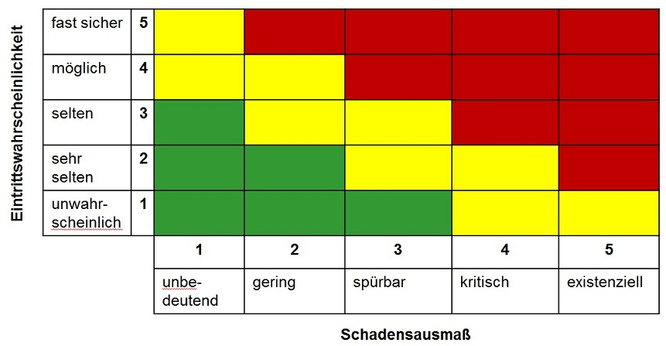
Abbildung 3: Risiko-Portfolio, Multiplikation der Klassen-Einstufung

Abbildung 4: Steuerung der Risiken, Steuerungsmaßnahmen
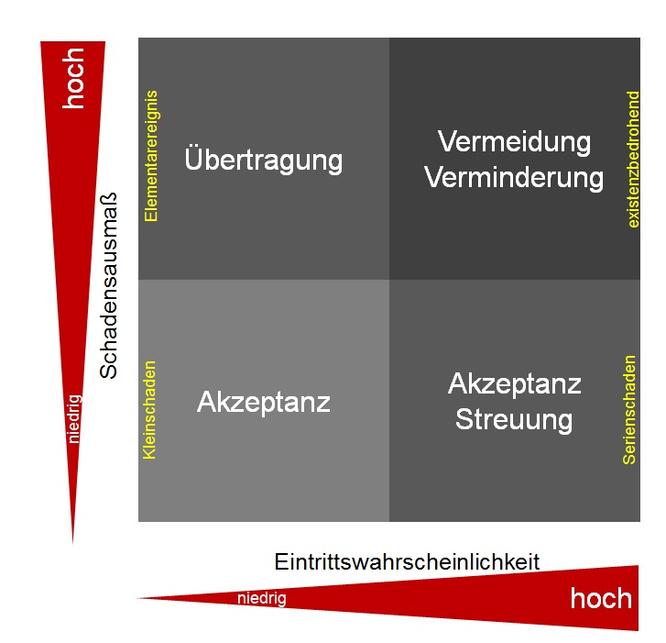
Abbildung 5: Steuerungsmaßnahme gemäß Risiko-Portfolio
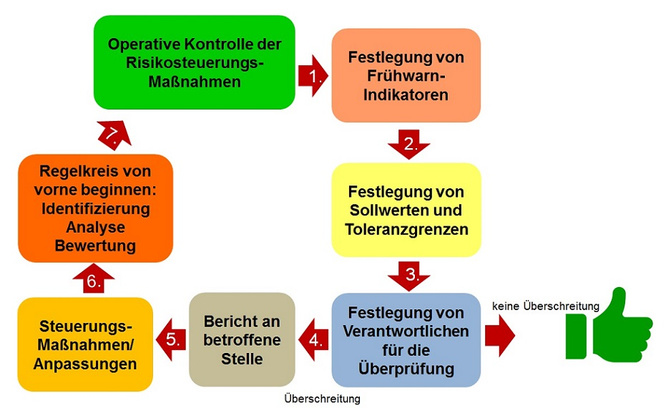
Abbildung 6: Bericht und Kontrolle der Risiken
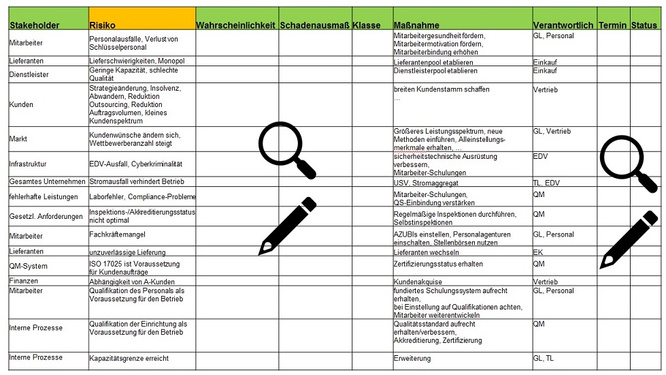
Abbildung 7: Beispielhafte Darstellung der Risikotabelle als to-do-Liste