Verifizierung von Arzneibuchmethoden im analytischen Labor

Unter folgendem Link können Sie den in der Pharmind (2) 2017 erschienen Artikel Verifizierung von Arzneibuchmethoden im analytischen Labor – wie umfangreich in der Praxis? als pdf einsehen.
Gerne unterstützen wir Sie bei Ihren Verifizierungen, sei es beratend oder durchführend.
Verifizierung von Arzneibuch-Methoden im analytischen Labor – Wie umfangreich in der Praxis?
Abstract
Die Verifizierung von chemisch-physikalischen Arzneibuchmethoden ist im Bereich der pharmazeutischen Analytik ein wichtiges Instrument zur Eignungsprüfung von bereits bestehenden Methoden aus den Pharmakopöen. Diese gelten als valide und müssen bei Anwendung demnach nicht einer erneuten Validierung unterzogen werden. Lediglich die Eignung unter den Umgebungsbedingungen des durchführenden Labors wird einer Prüfung unterzogen. Doch gerade die praktische Umsetzung steht oft vor vielen Fragezeichen, da Art und Anzahl der zu überprüfenden Parameter und der dazugehörigen Akzeptanzkriterien zur Erstellung eines individuellen Verifizierungsplans im Vorfeld regulatorisch nicht konkret vorgegeben sind. So summieren sich Fragen nach der Beurteilung einer Methode, den relevanten Verifizierungsparametern oder den Akzeptanzkriterien und letztendlich dem gesamten Umfang der Verifizierung, bevor eine geeignete Implementierung in die Routine erfolgen kann.
Einleitung
Die Methodenverifizierung stellt eine Teilvalidierung der Methode dar, bei der nur die für die jeweilige Methode zu den gegebenen Bedingungen kritischen Methodenparameter überprüft werden. Arzneibuchmethoden gelten per se als valide, sie müssen lediglich einer Verifizierung unterzogen werden. Bei jeder anderen validierten Methode muss ein Methodentransfer durchgeführt werden, wenn ein anderes Labor als das, das die Methode validiert hat, nach dieser prüft. In der Praxis stellt sich den meisten durchführenden Laboratorien nun die Frage nach der Art und dem Umfang der Verifizierung. Sie soll so umfangreich sein, dass alle kritischen Faktoren Beachtung finden, jedoch soll der Aufwand gleichzeitig möglichst gering gehalten werden. Im Folgenden werden mögliche Ansätze zur Ermittlung des Prüfumfangs, der Auswahl der Verifizierungsparameter und zu dem Setzen von Akzeptanzkriterien, sowie die Probleme, die damit einhergehen, erörtert.
Regulatorischer Hintergrund
Welche Regelwerke geben handfeste Vorgaben, mit deren Hilfe eine sinnvolle Verifizierung erfolgen und auf die sich berufen werden kann? Wer im europäischen Raum Arzneibuchmethoden verifizieren möchte und Handlungshilfen zur Durchführung des praktischen Eignungstests sucht, wird im europäischen Arzneibuch kaum fündig werden. Lediglich ein kurzer Abschnitt im Europäischen Arzneibuch (Ph. Eur.) weist darauf hin, dass eine komplette Validierung nicht erforderlich, aber eine Eignungsprüfung bzw. Verifizierung der Methode notwendig ist: „Validierung von Arzneibuch-Methoden: […] Falls in der Monographie oder dem Allgemeinen Kapitel nichts anderes vorgeschrieben ist, ist eine Validierung der Prüfmethoden nicht erforderlich.“ „Implementierung von Arzneibuch-Methoden: Wenn eine Arzneibuch-Methode eingesetzt wird, muss der Anwender evaluieren, ob und in welchem Ausmaß die Eignung der Methode […] nachgewiesen werden muss.“[1] Auch im GMP-Leitfaden Kapitel 6 ist beschrieben, dass das durchführende Labor die Eignung der Methode belegen muss, wenn es nicht die Validierung durchgeführt hat. Dies bedeutet für validierte Methoden, die nicht aus dem Arzneibuch stammen, einen Methodentransfer oder eine (Teil-)Validierung und für Arzneibuchmethoden die Durchführung einer Methodenverifizierung [2]. Lässt man den Blick weiter schweifen zu amerikanischen Regelwerken und Leitlinien wie der United States Pharmacopeia (USP) [3], dem Code of Federal Regulations (CFR) [4] oder der im Juli 2015 herausgegebenen FDA Guidance Analytical Procedures and Methods Validation for Drugs and Biologics [5] kann man einige Anhaltspunkte für relevante Parameter finden, um die Eignung der Methode im eigenen Labor zu belegen. Auch wenn es bei einer Prüfung nach europäischem Arzneibuch bzw. für den europäischen Markt nicht bindend ist, stellen diese Vorgaben eine gute Basis, auf die man sich berufen kann, dar. Wann eine Verifizierung und keine Validierung notwendig ist, wird deutlich beschrieben im CFR Laboratory Records. (21 CFR 211.194). Hier wird außerdem der geforderte Umfang, der an eine vollständige Dokumentation von Laboraufzeichnungen gestellt wird, definiert. Laut CFR muss hier ein Verweis auf die verwendete Methode erfolgen und der Nachweis, dass diese Methode auch geeignet bzw. validiert ist. Methoden aus der USP, dem NF (National Formulary) oder AOAC (Association of Analytical Communities) -Methoden müssen nicht vor Ort validiert sein. Es reicht ein einfacher Verweis aus. Aber alle Methoden sollen unter den jeweils aktuellen Umgebungsbedingungen verifiziert sein. Im USP Kapitel <1226> Verification of Compendial Procedures wird festgelegt, dass eine Verifizierung nicht das bloße Nachbilden einer Validierung bedeutet, sondern nur dazu verwendet wird Daten zu erheben, die die Eignung der Methode unter den jetzigen Umgebungsbedingungen aufzeigen. Der Umfang kann also von dem einer vollständigen Validierung nach ICH abweichen bzw. individuell festgelegt werden in Abhängigkeit von
- der Art der vorliegenden Methode,
- der Erfahrung des Analytikers mit einem solchen Prüfverfahren,
- den eingesetzten Geräten oder Ausrüstungen,
- der Probenaufarbeitung,
- der Art des Probenmaterials.
In der FDA Guidance Analytical Procedures and Methods Validation for Drugs and Biologics (July 2015) wird ebenfalls die Verifizierung von Arzneibuchmethoden gefordert. Die Guideline führt aus, dass die Verifizierung nach einem Verifizierungsplan mit festgesetzten Akzeptanzkriterien durchgeführt werden soll. Der Plan soll einen detaillierten Bezug zur Methode herstellen. Die Prüfung der Robustheit ist laut Guidance nicht notwendig, wenn keine Abweichungen von der Methode bestehen.
Welche Parameter geprüft werden, soll von der aktuellen Fragestellung abhängig gemacht werden. Dies kann beispielsweise bedeuten, dass bei Abweichung der internen Spezifikation von der Arzneibuchspezifikation, der Umfang der Verifizierung entsprechend erhöht werden muss, da nicht davon ausgegangen werden kann, dass die Methode per se für diesen Zweck geeignet ist. Bei Wirkstoffen, denen verschiedene Syntheserouten und somit unterschiedliche Verunreinigungsmuster zu Grunde liegen können, ist bei einer Reinheitsprüfung die Überprüfung einer möglichen Interferenz einzelner Peaks innerhalb eines Chromatogramms bzw. die Überprüfung der Selektivität sinnvoll und wichtig. Auch bei fertigen Darreichungsformen (FDFs), bei denen der Herstellungsprozess und die Matrix einen großen Einfluss besitzen, muss der Umfang ebenfalls ausgeweitet werden. Liegen diese Fälle nicht vor kann auch eine Verifizierung in einem sehr kleinen Umfang erfolgen. Wenn für den US-amerikanischen Markt geprüft wird, sollte beachtet werden, dass nur Methoden die in 21 CFR 211.194 als solche Methoden benannt sind, verifiziert werden müssen. Methoden aus dem europäischen Arzneibuch sind nicht explizit in der Verordnung benannt und müssen nach rein formeller Lage einer Validierung unterzogen werden, wenn sie für den US-amerikanischen Markt verwendet werden sollen.
Bewertung der Prüfmethode und Bewertung der Komplexität des Probenmaterials
Der erste Schritt im Rahmen der Festlegung des Verifizierungsumfangs ist die Bewertung der in der jeweiligen Monographie beschriebenen Methode. Dies beginnt mit der Betrachtung, ob es sich beispielsweise um eine Prüfung auf Identität, Gehalt, Reinheit, Wirkstofffreisetzung oder um eine einfache Standardmethode wie der Bestimmung des pH-Werts, der Säurezahl oder des Trocknungsverlusts handelt, welche laut USP nicht verifiziert werden muss. Bei einer Gehaltsmethode muss unterschieden werden, ob es sich um eine absolute Methode wie die Titration handelt, bei der ein hohes Maß an Präzision vorliegt, dafür vielleicht ein einhergehender Verlust der Spezifität in Kauf genommen werden muss, oder um eine relative Methode wie ein chromatographisches Verfahren, bei der von einer höheren Spezifität ausgegangen werden kann. Ebenso kann das Beispiel um die Reinheitsprüfung erweitert werden. Hier kann es sich um einen Limittest wie die Bestimmung von Restlösemitteln handeln oder um eine quantitative Methode, wie sie zur Bestimmung von verwandten Substanzen angewandt wird. So ist die Art der Methode schon ausschlaggebend für den Umfang der Verifizierung. Die Beschreibung von Arzneibuchmethoden ist meistens sehr knapp gehalten, so dass der Analytiker sich fragen muss, ob alle relevanten Methodenparameter ausreichend beschrieben sind oder ob der Interpretationsspielraum direkt Auswirkungen auf die Qualität der Analysenergebnisse hat. Dies betrifft insbesonder
- Probenaufarbeitungen
- Detektionsmethoden
- verwendete Materialien im Allgemeinen
- Lagerung und Probenzug
Die eigentliche Fülle an möglichen Einflussgrößen kann durch ein Ishikawa-Diagramm (Abbildung 1) veranschaulicht werden.
Auswahl der Verifizierungsparameter
Wie wählt man nun die relevanten Verifizierungsparameter aus?
In der USP gibt es ein längeres Kapitel zur Verifizierung von Arzneibuchmethoden, das gute Hilfestellung liefert und den Umfang einer solchen Verifizierung konkretisiert. Im Kapiteil USP<1226> Verification of Compendial Procedures wird auf das allgemeine Validierungskapitel USP <1225> Validation of Compendial Procedures hingewiesen, in dem alle relevanten Parameter gelistet sind und je nach Verfahren eine konkrete Auswahl an möglichen zu prüfenden Parametern vorgeschlagen wird (Tabelle 1; vgl. ICH Q2(R1) Validation of Analytical Procedures: Text and Methodology [6]). Da dies Empfehlungen für Validierungen sind, kann davon ausgegangen werden, dass der Umfang für Verifizierungen im Normalfall immer geringer als hier vorgegeben ausfällt. Der Verifizierungsumfang wird vorab durch den Verifizierungsplan festgelegt. Dieser kann bei Standardmethoden unter anderem als eine Art Formblatt vorliegen. Bei der Erstellung des Plans sollten im Vorfeld bestimmte Faktoren abgeklärt werden, um den letztendlich relevanten Prüfumfang zu ermitteln. Im Folgenden sind mögliche Punkte zur Abklärung des Verifizierungsumfangs genannt:
1. Überprüfung der Methode (Arzneibuch, Klassifizierung der Methode)
- Ist die Methode bereits etabliert?
- Handelt es sich um eine einfache Standardmethode?
- Ist die Herstellung und Stabilität aller Lösungen genau beschrieben?
- Sind alle notwendigen Geräteparameter gegeben?
- Ist die Art der Auswertung und Formel gegeben und korrekt dargestellt?
- Gibt es Punkte, die nicht dem Stand der Technik entsprechen?
2. Untersuchungsumfang
- Feste Parameter, die standardmäßig überprüft werden können, wenn keine Vorgaben/ Beurteilungen vorliegen bzw. keine einfache Standardmethode vorliegt.
- SST
- Selektivität (Reinheitsbestimmungen)
- Linearität
- Methodenpräzision
- Intermediärpräzision
- Standardstabilität
- Probenstabilität
Als wichtigste Prüfparameter werden die Bestimmung der Selektivität sowie das Bestehen des der Methode eigenen System suitability tests (SST) als obligatorisch angeführt. Die allgemeinen Kapitel der Pharmakopöen sollten bei der Prüfparameterwahl hier nicht außer Acht gelassen werden. Bei der Überprüfung des SST nach Ph. Eur. für chromatographische Methoden werden beispielsweise die Systemeignungsparameter aus dem Kapitel 2.2.46 Chromatographische Trennmethoden aus dem europäischen Arzneibuch herangezogen. Diese müssen bei jeder Bestimmung erfüllt werden. Analog hierzu werden bei Prüfungen nach USP die Systemeignungsparameter des USP Kapitel <621> Chromatography – System Suitability herangezogen. Zur Durchführung chromatographischen Methoden empfiehlt es sich, die Retentionszeit im Vorfeld zu bestimmen, den System Suitability Test (SST) und die Tendenz zu Verschleppungen oder die Stabilität der Sequenz zu prüfen. Es lohnt sich unter Umständen eine Überprüfung, ob die Systemeignungstests aus der Monographie und dem allgemeinen Kapitel ausreichend sind oder ob noch weitere Tests hinzugefügt werden müssen. Beispielsweise kann ein Kontrollstandard am Ende der Analytik, die Bestimmung der Höhe der Abweichung zweier parallel hergestellter Referenzstandards, eine doppelte Probenaufarbeitung oder auch eine Injektion mit einem Wirkstoffgehalt an der Detektionsgrenze oder die Herstellung eines Wirkstoffstandards mit 100% Gehalt bei Reinheitsmethoden zur Überprüfung der vollständigen Extraktion der Probe stattfinden. Bei Reinheitsbestimmungen (auch bei Limittests) empfiehlt sich eine Ermittlung des Limit of Detection (LOD) bzw. des Limit of Quantification (LOQ). Die Überprüfung der Bestimmungsgrenze ist unter anderem sinnvoll, damit zum Beispiel im Analysen-Zertifikat falls in der Probe keine Verunreinigungen detektiert werden, ein Reporting Limit angegeben werden kann. Eine Bestimmung der Selektivität bei chromatographischen Reinheitsmethoden erleichtert es später in der Routine bekannte Verunreinigungen zuzuordnen, da dem Analytiker Vergleichs-Chromatogramme vorliegen. In der USP sind auch Verfahren für Fertigarzneimittel beschrieben. Hier empfiehlt es sich, auch eine Wiederfindung von Verunreinigungen und Wirkstoff aus der Matrix zu ermitteln. Die Prüfungen der Richtigkeit und Wiederfindung sind nur nötig bei komplexer Matrix (z.B. bei Methoden für FDFs in der USP) oder bei komplexer Probenaufarbeitung wie z.B. bei einer Derivatisierungsreaktion oder aufwendiger Extraktion. Die Bestimmung der Linearität ist im Prinzip nicht nötig, trotzdem sollte bei einer hohen Probenkonzentration überprüft werden, ob noch im linearen Bereich des Detektors gearbeitet wird. Falls nicht ist eine Verringerung des Injektionsvolumens (HPLC/GC) nach Ph.Eur. 2.2.46 erlaubt. In diesem Fall sollte aber die Linearität mit überprüft werden. Vor der Durchführung der Verifizierung und der Erstellung des Verifizierungsplans sollten bei der Installation der Methode bereits kleinere Robustheitsprüfungen durchgeführt werden, um die Methode besser bewerten zu können. Solche können die Prüfung der Eignung von Verbrauchsmaterialien wie Filter oder das Extraktionsverhalten der Probe umfassen. Bei einer Bestimmung der Robustheit sollte aus pragmatischen Gründen die Stabilität der Lösungen überprüft werden, um später in der Routine die eventuell nicht notwendige, mehrfache frische Aufarbeitungen der Proben und Standards zu umgehen. Weiterführende Informationen zur Festlegung des Verifizierungsumfangs oder zum Equipment das bei der ursprünglichen Validierung der Arzneibuch-Methoden verwendet wurde, enthalten der Kommentar zum Europäischen Arzneibuch, die EDQM-Knowledge Database [7] sowie die Webseite der USP zu den bei den Validierungen verwendeten HPLC-Säulen [8].
Setzen von Akzeptanzkriterien
Nach der Auswahl der zu überprüfenden Parameter müssen hierfür geeignete Akzeptanzkriterien im Vorfeld der Prüfung festgelegt werden. Bei der Methodenverifizierung soll die Methodeneignung im eigenen Labor unter den aktuellen Gegebenheiten aufgezeigt werden, die einmal natürlich methodenimmanent ist und von den Produkteigenschaften wie zum Beispiel der Qualitätslage und der Produktspezifikation bzw. dem Anwendungszweck abhängt. Hier gilt es das geeignete Maß zu finden. Liegen die Akzeptanzkriterien zu eng, schließt man vielleicht geeignete Methoden aus, liegen sie zu weit, gerät man in Gefahr in der späteren Routine häufig OOS-Ergebnisse zu erhalten oder auch einfach auf Grund der hohen Schwankungsbreite Daten zu erzeugen, die eigentlich keine Aussagekraft besitzen. Dies kann am Beispiel der Bestimmung der Präzision einer Gehaltsmethode erläutert werden. Was bedeutet nun die Präzision und die weite der hier gesetzten Akzeptanzkriterien einer Methode im Hinblick auf die Produktqualität und die Spezifikation? Während bei der Bestimmung der Wiederfindung oder Linearität ein möglicher systematischer Fehler aufgedeckt wird, ist die Bestimmung der Präzision ein Maß für den zufälligen Fehler oder die Messunsicherheit. Abbildung 2 zeigt, wie schnell man bei einem wahren Messwert von 98,5 % bei einer Bestimmung des Gehalts ein Out-of-specification (OOS) Ergebnis produziert, das nur auf die Messunsicherheit zurückzuführen ist. Die Ergebnisse sind normalverteilt und bei entsprechend hoher Streuung liegen entsprechend viele Messwerte unterhalb des Lower Specification Limits (LSL). Dies gilt natürlich auch in umgekehrte Richtung wenn der wahre Wert im OOS-Bereich liegt und der gemessene Wert vermeintlich innerhalb der Spezifikation liegt. So sollten selbst bei weit gesetzten Akzeptanzkriterien für den Gehaltsprozentwert die Akzeptanzkriterien für die Streuung (relative Standardabweichung) nicht erweitert werden. Nun könnte man diesem Problem begegnen, indem die Probe aufgereinigt wird oder durch die Erweiterung der Spezifikation. Dies liegt jedoch meist nicht in der Macht des durchführenden Analytikers. Eher kann die Streuung bzw. die Messunsicherheit reduziert werden, auch wenn das Verfahren an sich nicht einfach geändert werden und im besten Fall durch proportionales Vergrößern der Proben- und Standardeinwaage eine höhere Genauigkeit erzeugt werden kann. Am einfachsten gelingt in den meisten Fällen eine drastische Reduzierung der Messunsicherheit, indem Mehrfachbestimmungen durchgeführt werden und die Akzeptanzkriterien für die relative Standardabweichung der Einzelwerte ausreichend eng, unabhängig von der Weite der Akzeptanzkriterien des Gehaltprozents, liegen. So kann auf Basis der durchgeführten Messung der Streuung bei guten Messergebnissen später eine Einfachbestimmung in der Routine vertreten werden. Analog sollte die Abwägung der Weite der Akzeptanzkriterien auf alle anderen einzelnen Parameter übertragen werden.
Mögliche Vorgehensweise und Akzeptanzkriterien, wenn das vorige Beispiel der Gehaltsbestimmung mittels HPLC aufgegriffen wird, sind, den SST der Monographie (Selektivität und Gerätepräzision (RSD ≤ 0,85%; n=6) )durchzuführen, wenn alle durch den Rohstoffhersteller ausgewiesenen Verunreinigungen durch die Monographie abgedeckt werden; zusätzlich die Durchführung der Präzision (RSD ≤ 2,0%) und fakultativ die Standard- und Probenstabilität (Abw. Zum Initialwert ≤ ± 2,0%). Die Linearität wird meist nicht überprüft. Sollte eine Überprüfung der Linearität notwendig sein, wird meist der Bereich 80 – 120 % verifiziert (Korrelation ≥ 0,999 / %y-Achsenabschnitt ≤ ± 3% / Darstellung: Residuenplot + Regressionsgerade / Bericht: Summe der Residuen² + Steigung). In diesem Fall könnte die Wiederfindung direkt über die Linearität bei matrixbehafteten Proben analytisch abgedeckt werden bzw. die Linearität umgekehrt aus den Daten der Richtigkeit oder auch anderen statistischen Methoden abgeleitet werden (vgl. USP <1210> Statistical Tools for Procedure Validation; Draft PF 40 (5)).
Setzen von Akzeptanzkriterien – Analytical Target Profile
Derzeit wird von Seiten der USP an einem Lebenszykluskonzept für Methoden gearbeitet, welches im neuen Kapitel <1220> The Analytical Procedure Lifecycle dargestellt werden soll. Hierzu erschienen auch mehrere „Stimuli“-Artikel, die sich mit der Erstellung eines sogenannten analytical target profiles (ATP) beschäftigen und auch Beispiele zur Formulierung solcher anführen [10], [11]. Das ATP beschreibt das genaue Ziel des Validierungsprozesses mit den relevanten Leistungsmerkmalen der Methode (engl. Method performance characteristics). Diese sollten entsprechend dem Ziel der Validierung sowohl zum Ausschluss von systematischen Fehlern, als auch zum Minimieren der Streuung der Werte ausgewählt werden. Das ATP wird als Text formuliert und ist nicht methodenspezifisch sondern produktspezifisch, wobei das Produkt in diesem Fall die zu erhaltenen Werte darstellt, welche durch die im ATP beschriebene Akzeptanzkriterien vorgegeben werden. So wird jeweils für die Bestimmung des Gehalts, Reinheit etc. ein eigenes ATP formuliert. Die Erstellung eines ATPs ist noch ein relativ junges Modell, dessen definiertes Ziel es ist, letztendlich durch genaue Richtlinien alle möglichen Streuungsquellen von Werten oder Messunsicherheiten zu berücksichtigen und zu minimieren, ohne nicht zielführende Vorgaben anzugeben und so durch den gesamten Lebenszyklus und somit bei der Verifizierung, angewendet werden kann.
Fazit
Die Herausforderung, den Spagat zwischen Aufwand und Nutzen bei der Verifizierung von Arzneibuchmethoden zu meistern, stellt viele Analytiker vor Herausforderungen. Hilfestellung bietet hier der Blick in die verschiedenen Regularien und die Ansätze, die darin enthalten sind, jedoch muss auch mit logischem Sachverstand jede Methode für sich betrachtet und der notwendige Umfang der Verifizierung ermittelt werden. Im Grenzfall sollte die Auswahl der Verifizierungsparameter umfangreicher sein, um Probleme in der Routine zu vermeiden. Durch ein vorzeitiges Auseinandersetzen mit der Art der Methode und den Akzeptanzkriterien für ein Produkt bzw. Ergebnis, können die zu erreichenden Ergebnisse und der zu erwartende Umfang der Analytik besser überblickt werden.
Quellen
[1] Europäisches Arzneibuch, 8. Ausgabe.
[2] Leitfaden der Guten Herstellungspraxis, Kapitel 6 (2014)
[3] U.S. Pharmacopeia & National Formulary, USP 39-NF 34.
[4] The Code of Federal Regulations of the United States of America, 21 CFR 211.194 (2016)
[5] Food and Drug Administration, Guidance Analytical Procedures and Methods Validation for Drugs and Biologics (July 2015): www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm386366.pdf (11/2016)
[6] International Council for Hamonisation of Technical Requirements for Pharmaceuticals for Human Use, Q2 Analytical Validation: www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (11/2016)
[7] EDQM Knowledge Database: extranet.edqm.eu/publications/recherches_sw.shtml (11/2016)
[8] USP Chromatographic Columns: www.uspchromcolumns.com (11/2016)
[9] B. Dejaegher et al., Improving method capability of a drag substance HPLC assay. Journal of Pharmaceutical and Biomedical Analysis 42 (2006) 155-170
[10] G.P. Martin et al., Stimuli to the Revision Process: Lifecycle Management of Analytical Procedures: Method Development, Procedure Performance Qualification, and Procedure Performance Verification. PF 39(5) (2016)
[11] K.L. Barnett et al., Analytical Target Profile: Structure and Application Throughout The Analytical Lifecycle. PF 42(5) (2016)
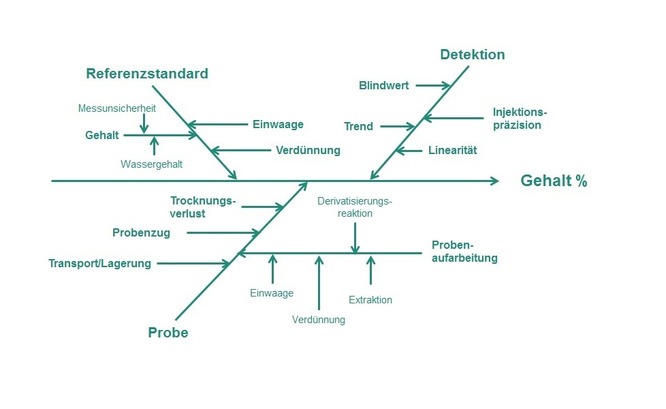
Abbildung 1: Ishikawa-Diagramm am Beispiel einer Gehaltsbestimmung mittels HPLC zur Darstellung kritischer Parameter und Einflussgrößen.
Tabelle Parameter Verifizierung
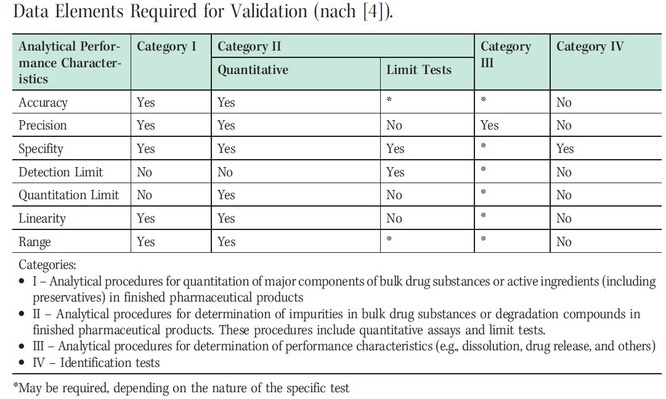
Tabelle 1: Data Elements required for Validation (nach [4])
Normalverteilung

Abbildung 2: Normalverteilung der Messwerte bei einem wahren Wert von 98,5%. Alle Werte innerhalb des Bereiches des Lower specification Limits (LSL) von 98,0 % und des Upper specification limits (USL) von 102,0 % liegen innerhalb der Spezifikation. Je näher der wahre Wert an einer der beiden Grenzen liegt, desto wahrscheinlicher wird es über die Gesamtheit der Werte ein OOS-Ergebnis zu erhalten [9].

